Research Philosophy
Our guiding philosophy is that twenty-first century biological research requires both “traditional” wet-lab as well as computational/bioinformatics approaches to studying the same set of problems. Hypotheses and ideas generated using one set of methods is tested and explored using the other, and lab members are encouraged to become conversant with both the test tube and the computer. The powerful combination of in silico and in vivo approaches enables us not only to make predictions but also to validate them within specific biological contexts.
Research Overview
The driving biological question in our laboratory is understanding the genetic regulatory circuitry that determines how cell fates are determined during development, with respect to both mechanism and evolution. We focus on two key aspects, intercellular signaling and transcriptional regulation, with the majority of our effort on the latter. Our focus is on the fruitfly Drosophila melanogaster due to its extremely well-annotated genome and amenability to experimental manipulation; the disease vector mosquito, Aedes aegypti; and other insects of medical and agricultural importance. We are successfully pioneering methods for studying the regulatory genomics of these diverse species. Our conclusions are also expected to relate directly to mammalian (including human) gene regulation.
Regulatory Networks
Gene expression is controlled by the binding of transcription factors to specific cis-regulatory elements. In the higher eukaryotes, these elements can lie 5' to, 3' to, or within introns of a gene; in some cases, they can even be found within protein coding sequences! Spatial and temporal aspects of gene expression are often controlled in a modular fashion, with individual cis-regulatory elements (termed "modules" or "enhancers") regulating expression in a particular time and place. An emerging theme is that a specific combination of transcription factors activiated as a result of intercellular signaling binds a regulatory module in conjunction with tissue-specific transcription factors ("selectors"), forming a "transcriptional code" that regulates the expression of a given gene (see Figure).
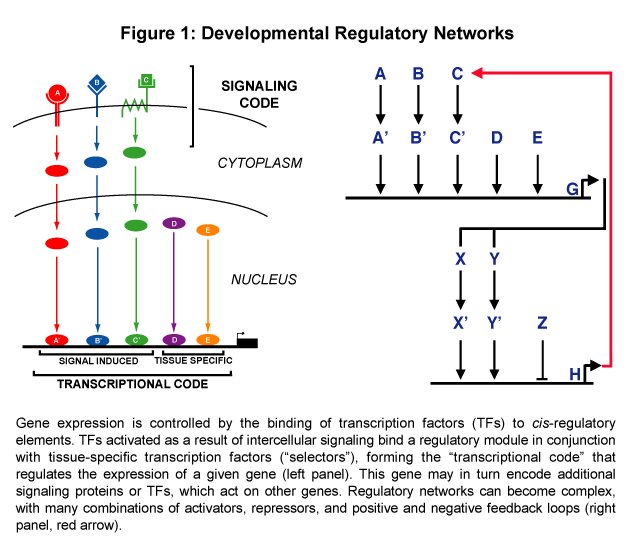 Together, the signaling and transcriptional events form a network of interactions in which signaling induces gene transcription, which can in turn lead to further signaling events, which then induce additional gene expression, and so on. Cascades of transcription can also occur, whereby transcription factors induce the expression of other transcription factors, which can in turn regulate still other transcription factors. These developmental regulatory networks are often complex, with multiple levels of cross-talk between different signaling pathways and both positive and negative feedback loops (see Figure 1). Our ultimate goal is to be able to describe all of the regulatory interactions involved in embryonic development. We are also interested in understanding how regulatory networks evolve.
Together, the signaling and transcriptional events form a network of interactions in which signaling induces gene transcription, which can in turn lead to further signaling events, which then induce additional gene expression, and so on. Cascades of transcription can also occur, whereby transcription factors induce the expression of other transcription factors, which can in turn regulate still other transcription factors. These developmental regulatory networks are often complex, with multiple levels of cross-talk between different signaling pathways and both positive and negative feedback loops (see Figure 1). Our ultimate goal is to be able to describe all of the regulatory interactions involved in embryonic development. We are also interested in understanding how regulatory networks evolve.
Defining cis-regulatory elements
cis-Regulatory modules (CRMs, "enhancers") are critical nodes in developmental regulatory networks, as it is here that signaling pathways and transcription factors are integrated to give rise to changes in the expression of specific genes. Mutations within CRMs have been implicated in a growing number of diseases, underscoring the importance of being able to identify and characterize them. We are using computational approaches to locate the cis-regulatory modules responsible for directing specific patterns of gene expression in a rapid and comprehensive fashion. All of our predictions are extensively tested in vivo using reporter gene assays in the fly embryo so that we can definitively assess our success rate and refine our approach to achieve better performance. SCRMshaw, our Supervised CRM discovery approach, has been developed in collaboration with Dr. Saurabh Sinha at the University of Illinois Urbana-Champaign and has proved to be highly effective for CRM discovery in both insects and mammals.
Importantly, we have demonstrated that SCRMshaw enables us to find CRMs across a wide range of insect species spanning over 345 million years of evolutionary divergence. We are using this ability to predict CRM sequences in the majority of sequenced insects of agricultural interest. In collaboration with Dr. Yoshinori Tomoyasu at Miami University, we are validating these predictions in both transgenic flies and transgenic beetles.
Genome Annotation
Much can be learned from studying already-known CRMs using bioinformatics approaches. For this reason, we have constructed the REDfly database of published Drosophila CRMs. This database contains more than 24,000 CRMs associated with over 1600 genes, along with their sequences and the expression patterns for which they are responsible. REDfly is an internationally-accessed resource that serves as a source of raw data for analysis, hypothesis generation, assessment and validation, and empirical research. As the only comprehensive, unbiased regulatory element database for any metazoan, it is widely used by researchers regardless of whether or not Drosophila is their model system of choice, and it has played a significant role in developing and validating methods that can be applied to vertebrate as well as insect systems. REDfly data have been important for studies of CRM biology, for interpretation of data obtained from genome-scale experiments, for facilitating both computational and empirical CRM discovery, for developing gene regulatory network models, and for studying CRM and regulatory network evolution.
Evolution of Gene Regulatory Networks (GRNs)
Changes in gene regulatory networks, which can occur at the level of CRMs or transcription factors, are important mediators of evolution. We have been investigating in detail a poorly-understood set of evolutionary mechanisms in which a gene regulatory network’s activity is shifted to a new tissue, and a different network appears to take over its role in the old tissue—a mode we refer to as “repeal, replace, and redeploy.” This research focuses on nervous system development in the embryos of mosquitoes (Aedes aegypti) and fruit flies (Drosophila melanogaster), taking advantage of two highly tractable research organisms with a mature selection of experimental tools, using our SCRMshaw software to identify the relevant gene regulatory sequences in each organism. The results will provide insight into how regulatory networks evolve and will build a foundation for in-depth functional analysis of the consequences of such evolution, over a greater evolutionary distance than has been possible in most previous studies. As an added impact, this work will lead to increased knowledge of the developmental biology of the Zika, dengue, and yellow fever vector mosquito Aedes aegypti and generate new tools and resources for the study and experimental manipulation of this important organism.
Signaling specificity in development and disease
Understanding intercellular signaling is an important component of defining the gene regulatory networks responsible for cell fate determination. We have been particularly interested in the various ways in which a class of signaling molecules knows as receptor tyrosine kinases (RTKs), mutations in which contribute to many birth defects and which are a major objective of targeted cancer therapeutics, produce unique downstream transcriptional responses. These receptors are often believed to be acting via identical downstream signaling cascades, but we have identified substantial points of divergence within the pathways (Leatherbarrow and Halfon, 2009). We have been focusing on the Drosophila visceral mesoderm (gut muscle), where the effects of RTK signaling appear to be mediated by a complex interplay of activators and repressors belonging to the ETS family of transcription factors. These studies will contribute to our understanding of RTK pathway regulation as well as identify additional genes important for mesoderm development.


